
")
Svelato il mistero sull’origine degli animali: le spugne sono le mamme di tutti (VIDEO)
Messo in discussione lo status quo accettato: i nostri primi antenati non sono gli ctenofori
www.greenreport.it
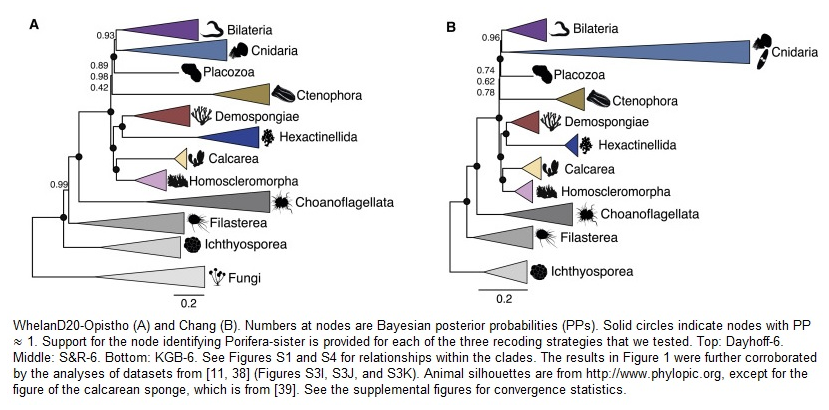
Il nuovo studio “Improved Modeling of Compositional Heterogeneity Supports Sponges as Sister to All Other Animals” pubblicato su Current Biology da in team di ricercatori dell’università di Bristol avrebbe risolto il più discusso mistero della biologia evolutiva, rivelando che il lignaggio più antico degli animali viventi è quello delle spugne, morfologicamente semplici, piuttosto che il phylum Ctenophora anatomicamente complesso. A capovolgere a favore delle spugne la convinzione che gli ctenofori fossero i nostri remoti antenati erano state recenti analisi genomiche, ma alcuni scienziati suggerivano cautela, perché i dati disponibili potrebbero non bastavano a risolvere il mistero. Ma la nuova ricerca condotta dall’Università di Bristol ha identificato la causa di questo effetto “flip-flop” e, così facendo, ha rivelato che le spugne sono il lignaggio più antico.
A questo risultato si è arrivati dopo che Davide Pisani, della School of biological sciences e della school of Earth sciences dell’Università di Bristol, e il suo team di ricerca formato da Roberto Feuda del California Institute of Technology (Caltech), Martin Dohrmann e Gert Wörheide della Ludwig-Maximilians-Universität (LMU), Walker Pett della Iowa State University, Hervé Philippe e Nicolas Lartillot del Cnrs, Omar Rota-Stabelli del centro di trasferimento tecnologico Fondazione Edmund Mach (FEM) di San Michele all’Adige (TN), hanno analizzato tutti i principali dataset genomici pubblicati tra il 2015 e il 2017.
Commentando questa svolta nella ricerca, Pisani ha spiegato: «Il fatto è che le ipotesi su chi sia venuto prima tra spugne o ctenofori suggeriscono storie evolutive completamente diverse per i principali sistemi degli organismi animali come il sistema nervoso e il sistema digestivo. Pertanto, conoscere il corretto ordine di ramificazione alla radice dell’albero [evolutivo]degli animali è fondamentale per comprendere la nostra evoluzione e l’origine delle caratteristiche chiave dell’anatomia animale».
Nel nuovo studio, Pisani e i suoi colleghi hanno utilizzato tecniche statistiche all’avanguardia come le posterior predictive analyses «per verificare se i modelli evolutivi utilizzati abitualmente nella filogenetica possano descrivere adeguatamente i dataset genomici utilizzati per studiare la prima evoluzione animale» e hanno scoperto che «Per lo stesso dataset, i modelli che possono descrivere meglio i dati favoriscono le spugne alla radice dell’albero degli animali, mentre i modelli che non riescono a descrivere drasticamente i dati favoriscono gli ctenofori».
Feuda aggiunge: «I nostri risultati offrono una semplice spiegazione dell’”effetto flip-flop “discusso in modo convincente dal professor David Hillis in una recente intervista su Nature». Secondo Dohrmann, «I nostri risultati razionalizzano questo effetto e illustrano come trarre solide conclusioni da dataset flip-flopping».
Wörheide fa notare che «In effetti, se analizzato utilizzando diversi modelli evolutivi, un dataset flip-flopping è un insieme di dati che supporta diverse storie evolutive o alberi filogenetici. Discriminare tra ipotesi alternative di fronte a un dataset flip-flopping richiede di chiarire quanto siano validi i modelli che supportano degli alberi filogenetici alternativi. Le posterior predictive analyses ci permettono di fare esattamente questo. Abbiamo scoperto che i modelli che descrivono dati scarsi identificano invariabilmente gli ctenofori alla radice dell’albero. I modelli che descrivono meglio i dati trovano invariabilmente le spugne in quella posizione».
Pisani conclude: «La filogenomica, l’utilizzo dei dati genomici nella filogenetica, è una scienza relativamente nuova. L’evidenza degli ctenofori come il più antico lignaggio della ramificazione degli animali è emersa per la prima volta nel 2008, una decina di anni fa, nella prima analisi filogenomica su larga scala dei phyla animali. Ora abbiamo strumenti e dati analitici migliori e questo studio mette seriamente in discussione lo status quo accettato».

